There are flaws in PROBABILITY ZERO. There are mistakes. There aren’t very many, to be sure, but there are a few. That’s why I’m working on the second edition now, to address those little flaws and mistakes, and to bring the book up-to-date with the very latest scientific studies. At Reddit, a number of the regulars on the r/DebateEvolution have collectively assembled a 333-comment thread to refute a book that none of them have read. This is, of course, the safest way to refute a book if one is primarily concerned with convincing oneself instead of anyone who has actually read it. The critiques come in a recognizable pattern, as each objection sounds authoritative and self-assured, and each one collapses the moment it is checked against what the book actually says and the available scientific evidence.
Objection 1: “How does Day deal with multi-base-pair mutations? ERVs, gene duplications, LINEs, SINEs, indels — does he count those as single events or as hundreds of thousands of mutations each?”
This is the most substantive question in the thread, which is presumably why it’s the one that inspires the least engagement. The answer is that it doesn’t matter.
In Yoo et al. (2025) the complete telomere-to-telomere assemblies of all great ape genomes are published. The Yoo numbers give us approximately 35 million single-nucleotide variants on the human lineage, plus 1,140 interspecific inversions, plus ~187 Mb of structurally divergent sequence. Total: about 205 million genomic differences requiring explanation.
Now, the critic’s excuse is to say “but inversions and structural variants are single events, not millions of mutations.” Fine. Discount every structural variant in the Yoo data to zero. Count nothing but single-nucleotide variants. The shortfall on the SNV-only subset is still four to five orders of magnitude. Going the other direction — counting every base pair in every structural variant as a separate mutation — pushes the shortfall to six orders of magnitude. The conclusion holds either way. Counting structural variants as single events is the maximally generous treatment, and the model still fails.
- Full Yoo et al. data: ~410 million total human-chimp differences → ~205 million apportioned to the human lineage → shortfall of ~1.1 × 10⁶, six orders of magnitude at 1/1,100,000.
- SNV-only, most conservative: ~17.5 million SNVs on the human lineage → shortfall of ~9.4 × 10⁴, nearly five orders of magnitude at 1/94,000.
The shortfall got worse by an order of magnitude when complete telomere-to-telomere assemblies replaced the older Chimpanzee Genome Project numbers. That’s the opposite of what one would expect if my original argument were based on cherry-picked or out-of-date inputs.
The critic was essentially asking, “did you put your finger on the scale in favor of evolution or against it?” The answer is: I calculated it iin favor of evolution, and evolution loses anyway.
Objection 2: “Whole genome duplication! Teleosts! Goldfish! Vertebrates have at least two rounds of ancient WGD!”
This is one of those objections where someone reaches for the heaviest object on the shelf without checking what’s actually inside the book. Yes, whole genome duplications happen. Yes, they’re real evolutionary events. They are also, however, totally irrelevant to the throughput argument, for three reasons that the critic didn’t consider.
First, a WGD doesn’t escape the fixation problem, but it intensifies it. A polyploidy event is a massive structural disruption that creates immediate compatibility problems with the rest of the breeding population. The standard outcome is sterility or inviability, not a new species. When it does succeed (mostly in plants, sometimes in fish), it succeeds via reproductive isolation of a tiny founding population. This means it’s a bottleneck speciation event, not a gradualist one. Polyploidy speciation has been observed precisely because it doesn’t operate by gradual substitution. It’s the opposite of the very mechanism the critic is trying to defend.
Second, the duplicated genes don’t automatically neofunctionalize. They have to mutate, and one of the two copies has to be silenced or repurposed, while the other continues doing its original job. The book explains the methylation and chromosomal-inactivation machinery required to shut down duplicate genes, a process that is itself complex and that has to be coordinated. You don’t get free new genes by doubling the genome. You get redundant, overproducing copies that immediately need to be regulated or eliminated.
Third, and most importantly, the Teleost-specific WGD doesn’t address the human-chimpanzee divergence problem. The consensus CHLCA is 6.3 million years ago, not 350 million years ago. No one is claiming the human lineage underwent a whole-genome duplication since splitting from chimpanzees. Pointing at fish from 350 million years ago to explain why ape divergence math works is the evolutionary-biology equivalent of explaining your tax shortfall by mentioning that someone, somewhere, won the lottery back in 1987.
Objection 3: “Sixteen papers haven’t overturned population genetics. None have been adopted by evolutionary biology. None have forced a textbook revision.”
This isn’t an argument. It’s an appeal to institutional inertia dressed up as an argument. Translated: the gatekeepers haven’t waved the white flag yet, therefore the gatekeepers are right.
Anyone who has paid attention to academic biology in the last twenty years knows what the peer review system actually rewards and punishes. The reproducibility crisis is now openly acknowledged in the literature, including, in Nature itself. Writing PROBABILITY ZERO led directly to a subsequent book on the structural problems that produce garbage science; HARDCODED even provides estimates of how much of every given field is already garbage and how long it will be before the still-functioning fields degrade entirely.
So the relevant question is not “have the papers forced a textbook revision?” The relevant question is: can anyone show that the math is wrong? The papers report a calculation. The inputs are the empirically measured fastest fixation rate ever observed (1,401 generations per fixation, Good et al. 2017, confirmed at whole-genome resolution by Couce et al. 2024). The outputs are arithmetic. If the calculation is wrong, the critics need to show where. None of them does. None of them even tries. They just appeal to the erroneous institutional consensus and call it refutation.
Stanislaw Ulam raised this same objection at the 1966 Wistar symposium. Sixty years later, the biologists still haven’t produced an answer. They can’t, because the math proves them wrong.
Objection 4: “He models evolution as a one-step random assembly problem instead of a cumulative, path-dependent, selection-filtered process.”
This is a flat misrepresentation, and a particularly lazy one, because the book is explicitly about cumulative fixation events at the fastest empirically observed rate. We are not calculating the probability of assembling a human genome in one shot. That’s Hoyle’s tornado-in-a-junkyard argument, and it isn’t even one of the many arguments in the book.
The argument in the book is this: take the fastest fixation rate ever measured in any organism — 1,401 generations per beneficial fixation in the E. coli long-term evolution experiment — and divide the time available since the human-chimpanzee divergence by that rate. You get approximately 186 fixation events on the human lineage. Then count the fixations required to account for the observed divergence. You need somewhere between 17.5 million (SNVs only, most generous count) and 205 million (full Yoo et al. divergence). The ratio of required to achievable is somewhere between 94,000 and 1.1 million.
This is not a one-step random assembly calculation. It is a cumulative throughput calculation using empirical fixation rates published by mainstream researchers in mainstream journals. The critic has invented a strawman to attack because the actual argument is impossible to dismiss.
Objection 5: “The ‘no ecologist has refuted it’ line is fantasy. Scientists don’t refute every bad argument. Silence is triage, not concession.”
Convenient. Also testable. If the argument can be refuted, it can be refuted. The math is published, the inputs are sourced from mainstream papers, and the calculation is elementary. Anyone who could show that 1,401 generations per fixation is wrong, or that more generations are involved, or that the divergence count is wrong, or that the arithmetic is wrong, would have an easy career-defining publication.
If evolutionary biologists could prove the mathematical possibility of evolution by natural selection, or even by natural selection and neutral theory, they would. They haven’t, they don’t, they can’t, and they won’t.
What’s actually happens is that the few evolutionary biologists who don’t simply run away from the subject concede the relevant inputs and then retreat to mechanisms that either don’t exist or don’t apply, or are insufficient to make their case. Triage is what you do when a problem is unworthy of engagement. But the people who engage are forced to concede the inputs. That’s not triage. That’s silence in the face of defeat.
Objection 6: “AI models don’t ‘reluctantly admit’ anything. They pattern-match text. User-induced hallucination dressed up as validation.”
This is the funniest one, because it shows that critic doesn’t understand how I utilize AI even though I’ve published a book explaining precisely that. Athos is listed as co-author on most of the technical papers. The role isn’t peer review; it’s calculation, formalization, and literature retrieval. The math either works or it doesn’t, and if the critics think Athos has been manipulated into producing false arithmetic, they are welcome to find the arithmetic error. They haven’t, because the arithmetic is correct. Note also that this objection is essentially “your tools are unreliable, therefore your conclusions are wrong.” This is not how science works. Galileo’s telescope was a tool. The objection isn’t to the tool; it’s to the conclusion. If you can’t show the conclusion is wrong, complaining about the tool is just venting.
Objection 7: “We have never witnessed speciation is flatly false. Speciation has been observed in plants, insects, fish, microbes, and laboratory populations.”
This requires unpacking what the critic is actually claiming. The book addresses speciation in detail and distinguishes between the categories of events the critic is collapsing together.
- Polyploidy in plants is genome duplication, not gradualist substitution. It is a single-event reproductive isolation mechanism that bypasses the Darwinian model. It is observed precisely because it doesn’t require millions of fixations. Citing polyploidy as an example of gradualist speciation is a category error.
- Ring species document partial reproductive isolation in progress over geological timescales. They are not complete speciation events observed in real time.
- Laboratory experiments in Drosophila and other organisms produce partial reproductive isolation under artificial selection. The isolation typically reverses when selection is relaxed. This is consistent with what the book predicts: micro-scale change within mathematical limits, full-scale speciation outside them.
The book’s quantitative claim, formalized in the Expected Speciation Frequency paper, is that if Darwinian gradualism worked as claimed, we should observe roughly 33 speciation events per year worldwide — one every eleven days. The observed rate of gradualist speciation in 3,000 years of recorded human observation is essentially zero. Polyploidy, ring species, and partial lab isolation don’t fill the gap. They are the rare exceptions the gradualist model cannot explain because they aren’t gradualist.
Objection 8: “Fruit flies and bacteria, evolution denial’s favorite props, have demonstrated novel traits, reproductive isolation, genomic divergence, and adaptive radiations.”
We agree they have demonstrated genomic divergence. So we ran the numbers on them. Drosophila melanogaster diverging from D. simulans, with the shortest generation time of any model animal: a shortfall factor of approximately 95. The fruit fly fails by two orders of magnitude.
Bacteria, on the other hand, pass the throughput test by a margin of more than a thousand. The book is explicit about this. Bacteria pass because they have no recombination delay, complete generational turnover (d ≈ 1.0), and astronomical generation counts in geological time. They are the only group that passes, and they pass because they lack the constraints that doom every sexual lineage.
Citing bacteria as evidence that the math works for sexual reproduction is like citing a fish as evidence that mammals can breathe underwater.
Objection 9: “Vox scales mutations per generation by generation time and stops there. He’s missing genome size and cell divisions per generation. He’s out by five orders of magnitude.”
This is the objection that initially sounds technical and substantive but turns out to be a confused conflation of two different quantities. The “5 orders of magnitude” math critique is confused in precisely the same way that Dennis McCarthy got it wrong, since it’s just another conflation of the mutation rate with fixation rate.
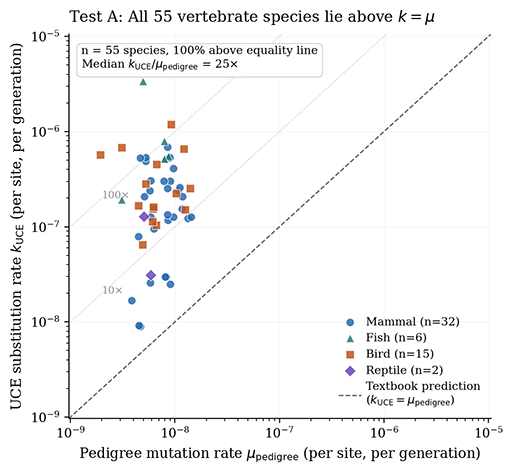
For some reason, many evolutionists somehow can’t understand the difference between one mutation occurring for the first time in a single individual and one mutation fixating across the billions of individuals that make up the species. But k does not equal u, fixation is a tiny subset of mutation, and it is a massive category error to confuse the two. The 100 mutations per individual per generation already incorporates genome size and germline cell divisions by definition. The bottleneck isn’t mutational occurrence, it’s mutational fixation.
Objection 10: “Mutations fix in parallel, not series. Each of those 20 million mutations could be fixing at the same time. Sixty mutations per generation × 450,000 generations = 21 million fixed mutations. Those are exceedingly reasonable numbers.”
This is the central rhetorical move that the entire chapter on parallel fixation in the book is designed to address.
Parallel mutation is real. Parallel fixation is not. The constraint is Haldane’s reproductive ceiling: the sum of selection coefficients across all simultaneously selected mutations cannot exceed what the population can bear in selective deaths per generation. Mathematically, Σsᵢ ≤ s_max. Try to select for one hundred beneficial mutations simultaneously, each with s = 0.01, and you’ve allocated a total selective load of 1.0 — meaning you’re killing the entire reproductive surplus of the population every generation. That’s extinction, not evolution.
Worse, Hill-Robertson interference makes parallel selection less efficient than serial selection. When multiple beneficial mutations segregate in the same population, they compete with each other for fixation. Ralph and Coop demonstrated in 2010 that this produces “soft sweeps” rather than the clean fixation events the standard model assumes.
The “60 mutations per generation × 450,000 generations = 21 million” calculation is what you get when you assume independent fixation of every mutation, with no reproductive constraint, no Hill-Robertson interference, no recombination limits, and no biological reality. It’s a back-of-the-envelope number that violates Haldane’s constraint by orders of magnitude. Reasonable, it is not.
This is also, incidentally, the same point to which JFG retreated to in our debate. He conceded the point about reproductive constraint only after I pressed him repeatedly. The defense doesn’t survive contact with the actual mathematics.
Objection 11: “A chromosome fusion: counted as a single mutation correctly, or wrongly as hundreds of thousands of individual mutations?”
Either way the model fails. Counted as a single event, you still need it to fix, and chromosome fusions create immediate meiotic incompatibility with the rest of the population, which makes fixation in a stable population effectively impossible. The human chromosome 2 fusion event is one of the standard cases the gradualist model has no good story for. Counted as many events, the throughput requirement explodes.
Structural variants and chromosomal rearrangements are worse for the gradualist model than point mutations, not better, because they break compatibility with non-carriers and therefore impede their own spread.
Objection 12: “Mutations fix faster during genetic bottlenecks. We know of at least a few extreme human ones.”
True, and the book uses the consensus effective population size of 10,000, which is already a bottleneck-adjusted figure; we’ve since calculated that the actual aDNA figure is 3,300. Going smaller helps fixation in two ways and hurts in three. It helps because drift-driven fixation is faster in smaller populations and because beneficial mutations have an easier time sweeping. It hurts because (a) smaller populations produce fewer novel mutations per generation, (b) smaller populations are subject to Muller’s ratchet — accumulating deleterious mutations faster than they can be purged — and (c) smaller populations are at higher risk of mutational meltdown and extinction.
The drift catastrophe is a serious problem, documented in the work of Kondrashov, Lynch, and Crow. Crow estimated that humans experience a 1-2 percent decline in genetic fitness per generation due to mutation accumulation. Bottleneck speciation gives you faster fixation at the cost of accelerated genetic decay. You can’t run that engine for 6.3 million years.
The Failure of the Redditors
Each individual objection sounds vaguely plausible if you don’t understand it. None of them survives even rudimentary examination. The pattern is consistent: the critics have constructed a version of the book they can refute, instead of engaging with the version that exists. They attack a one-step random assembly model the book doesn’t use. They cite parallel fixation calculations that violate Haldane’s constraint. They wave at speciation events that bypass the Darwinian mechanism. They invoke whole genome duplications that don’t apply to the ape lineage. They appeal to the institutional consensus and call it refutation.
The book’s central claim is arithmetic. Either the fastest empirically measured fixation rate, applied across the available time, can produce the observed divergence — or it can’t. The arithmetic says it can’t, by four to six orders of magnitude depending on how generously you count.
The Reddit critics haven’t shown the arithmetic of PROBABILITY ZERO is wrong. They’ve only shown they don’t want to do the math themselves.
DISCUSS ON SG