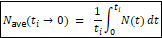A commenter at Dennis McCarthy’s site, John Sidles, attempts to refute my demonstration that Mooto Kimura made a fatal mistake in his neutral-fixation equation “k=μ”.
VOX DAY asserts confidently, but wrongly, in a comment:
“Kimura made a mistake in the algebra in his derivation of the fixation equation by assigning two separate values to the same variable.”
It is instructive to work carefully through the details of Kimura’s derivation of the neutral-fixation equation “k=μ”, as given in Kimura’s graduate-level textbook “The Neutral Theory of Molecular Evolution” (1987), specifically in Chapter 3 “The neutral mutation-random drift hypothesis as an evolutionary paradigm”.
The derivation given in Kimura’s textbook in turn references, and summarizes, a series of thirteen articles written during 1969–1979, jointly by Kimura with his colleague Tomoko Ohta. It is striking that every article was published in a high-profile, carefully-reviewed journal. The editors of these high-profile journals, along with Kimura and Ohta themselves, evidently appreciated that the assumptions and the mathematics of these articles would be carefully, thoroughly, and critically checked by a large, intensely interested community of population geneticists.
Even in the face of this sustained critical review of neutral-fixation theory, no significant “algebraic errors” in Kimura’s theory were discovered. Perhaps one reason, is that the mathematical derivations in the Kimura-Ohta articles (and in Kimura’s textbook) are NOT ALGEBRAIC … but rather are grounded in the theory of ordinary differential equations (ODE’s) and stochastic processes (along with the theory of functional limits from elementary calculus).
Notable too, at the beginning of Chapter 3 of Kimura’s textbook, is the appearance of numerical simulations of genetic evolution … numerical simulations that serve both to illustrated and to validate the key elements of Kimura’s theoretical calculations.
As it became clear in the 1970s that Kimura’s theories were sound (both mathematically and biologically), the initial skepticism of population geneticists eolved into widespread appreciation, such that in the last decades of his life, Kimura received (deservedly IMHO) pretty much ALL the major awards of research in population genetics … with the sole exception of the Nobel Prizes in Chemistry or Medicine.
Claim 1: My claim that Kimura made a mistake in the algebra in his derivation of the fixation equation by assigning two separate values to the same variable” is “confidently, but wrongly” asserted.
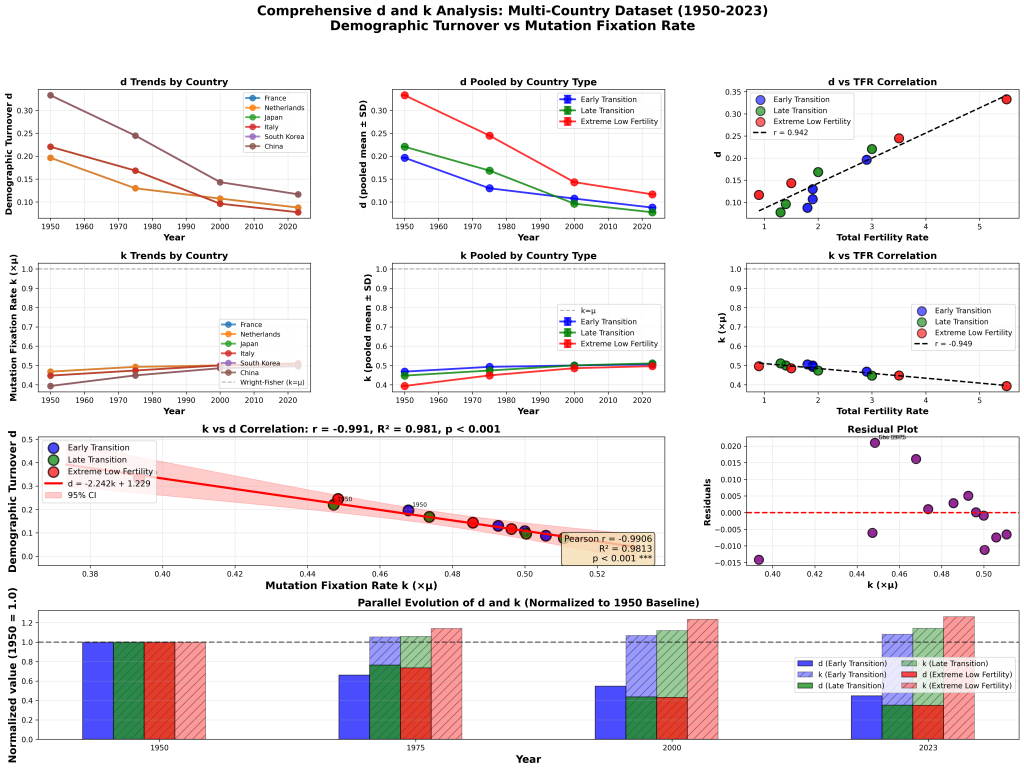
No, my claim is observably correct. The k = μ derivation proceeds in three steps:
Step 1 (mutation supply): In a diploid population of size N, there are 2N gene copies, so 2Nμ new mutations arise per generation. Here N is the census population—individuals replicating DNA. Kimura’s own 1983 monograph makes this explicit: “Since each individual has two sets of chromosomes, there are 2N chromosome sets in a population of N individuals, and therefore 2Nv new, distinct mutants will be introduced into the population each generation” (p. 44). This is a physical count of bodies making DNA copies.
Step 2 (fixation probability): Each neutral mutation has fixation probability 1/(2N). This result derives from diffusion theory under Wright-Fisher model assumptions, where N is the effective population size—the size of an idealized Wright-Fisher population experiencing the same rate of genetic drift. Kimura himself uses N_e notation for drift-dependent quantities elsewhere in the same work: “S = 4N_e s, where N_e is the effective population size” (p. 44).
Step 3 (the “cancellation”): k = 2Nμ × 1/(2N) = μ.
The cancellation requires the N in Step 1 and the N in Step 2 to be the same number. They are not. Census N counts replicating individuals. Effective N_e is a theoretical parameter from an idealized model. In mammals, census N exceeds diversity-derived N_e by ratios of 10× to 46× (Frankham 1995; Yu et al. 2003, 2004; Hoelzel et al. 2002). If the two N’s are not equal, the correct formulation is:
k = 2Nμ × 1/(2N_e) = (N/N_e)μ
This is not a philosophical quibble. It is arithmetic. If you write X × (1/X) = 1, but the first X is 1,000,000 and the second X is 21,700, you have not performed a valid cancellation. You have performed an algebraic error. The fact that the two quantities could be equal in an idealized Wright-Fisher population with constant size, random mating, Poisson-distributed offspring, and discrete non-overlapping generations does not save the algebra when applied to any natural population, because no natural population satisfies these conditions.
Claim 2: The derivation references thirteen articles published in “high-profile, carefully-reviewed journals” and was subjected to “sustained critical review” by “a large, intensely interested community of population geneticists.”
This is true and it is irrelevant. The error was not caught because the notation obscures it. When you write 2Nμ × 1/(2N), the cancellation looks automatic—it appears to be a trivial identity. You have to stop and ask: “Is the N counting replicating bodies the same quantity as the N governing drift dynamics in a Wright-Fisher idealization?” The answer is no, but the question is invisible unless you distinguish between census N and effective N_e within the derivation itself.
Fifty years of peer review did not catch this because the reviewers were working within the same notational framework that obscures the distinction. This is not unusual in the history of science. Errors embedded in foundational notation persist precisely because every subsequent worker inherits the notation and its implicit assumptions. The longevity of the error is not evidence of its absence; it is evidence of how effectively notation can conceal an equivocation.
John Sidles treats peer review as a guarantee of mathematical correctness. It is not, and the population genetics community itself has acknowledged this in other contexts. The reproducibility crisis affects theoretical as well as empirical work. Appeals to the number and prestige of journals substitute sociological authority for mathematical argument.
Claim 3: “No significant ‘algebraic errors’ in Kimura’s theory were discovered.”
This is an argument from previous absence, which is ridiculous because I DISCOVERED THE ERROR. No one discovered the equivocation because no one looked for it. The k = μ result was celebrated as an elegant proof of population-size independence. It became a foundational assumption of neutral theory, molecular clock calculations, and coalescent inference. Questioning it would have required questioning the framework that built careers and departments for half a century.
Moreover, the claim that no errors were discovered is now empirically falsified. I demonstrated that the standard Kimura model, which implicitly assumes discrete non-overlapping generations and N = N_e, systematically overpredicts allele frequencies when tested against ancient DNA time series. The model overshoots observed trajectories at three independent loci (LCT, SLC45A2, TYR) under documented selection, and a corrected model reduces prediction error by 69% across all three. A separate analysis of 1,211,499 loci comparing Early Neolithic Europeans with modern Europeans found zero fixations over seven thousand years—against a prediction of dozens to hundreds under neutral theory’s substitution rate.
The error has now been discovered. The fact that it was not discovered sooner reflects the fundamental flaws of the field, not the soundness of the mathematics.
Claim 4: The mathematical derivations “are NOT ALGEBRAIC… but rather are grounded in the theory of ordinary differential equations (ODE’s) and stochastic processes.”
This is true of Kimura’s fixation probability formula, P_fix = (1 − e^(−2s)) / (1 − e^(−4N_e s)), which derives from solving the Kolmogorov backward equation—a genuine boundary-value problem for an ODE arising from the diffusion approximation to the Wright-Fisher process. The commenter is correct that this piece of Kimura’s mathematical apparatus is grounded in sophisticated mathematics and is INTERNALLY consistent.
But it is not externally consistent and the k = μ result does not come from the ODE machinery anyhow. It comes from the counting argument: 2Nμ mutations per generation × 1/(2N) fixation probability = μ. This is multiplication. The equivocation is in the multiplication, not in the diffusion theory. Invoking the sophistication of Kimura’s ODE work to defend a three-line counting argument is a red herring. Mr. Sidles is defending Kimura on ground where Kimura is correct (diffusion theory) while the error sits on ground where the math is elementary (the cancellation of two N terms that represent different quantities).
The distinction between census N and effective N_e is not a subtlety of diffusion theory. It is visible to anyone who simply asks what the symbols mean. Mr. Sidles’s invocation of ODEs and stochastic processes does not address the actual error.
Claim 5: Numerical simulations “serve both to illustrate and to validate the key elements of Kimura’s theoretical calculations.”
Numerical simulations of the Wright-Fisher model validate Kimura’s results within the Wright-Fisher model. This is unsurprising—if you simulate a constant-size population with discrete generations, random mating, and Poisson reproduction, you will recover k = μ, because the simulation satisfies the assumptions under which the result holds.
The question is not whether Kimura’s math is internally consistent within its model. It is. The question is whether the model’s assumptions map onto biological reality. They observably do not. No natural population has constant size. No natural population of a long-lived vertebrate has discrete, non-overlapping generations. Census population systematically exceeds effective population size in every mammalian species studied.
Simulations that assume the very conditions under which the cancellation holds cannot validate the cancellation’s applicability to populations that violate those conditions. This is circular reasoning: the model is validated by simulations of the model.
Ancient DNA provides a non-circular test. When the standard model’s predictions are compared to directly observed allele frequency trajectories over thousands of years, the model fails systematically, overpredicting the rate of change by orders of magnitude. This empirical failure cannot be explained by simulation results that assume the model is correct.
Summary: Mr. Sidles’s defense reduces to three arguments: (1) many smart people reviewed the work, (2) the math uses sophisticated techniques, and (3) simulations confirm the theory. None of these address the actual error.
The error is simple: the k = μ derivation uses a single symbol for two different quantities—census population size and effective population size—and cancels them as if they were identical. They are not identical in any natural population. The cancellation fails.
The result that substitution rate is independent of population size holds only in an idealized Wright-Fisher population with constant size, and is not a general law of evolution.
Kimura’s diffusion theory is internally consistent within the Wright-Fisher framework and only within that framework. His fixation probability formula follows validly from its premises—premises that no natural population satisfies, since N_e is not constant, generations are not discrete, and census N ≠ N_e in every species studied. His contributions to population genetics are substantial.
None of this changes the fact that the k = μ derivation contains an algebraic error that has propagated through nearly sixty years of molecular evolutionary analysis.
In spite of this, Mr. Sidles took another crack at it:
Vox, your explanation is so clear and simple, that your mistake is easily recognized and corrected.
THE MISTAKE: “Step 2 (fixation probability): Each neutral mutation has fixation probability 1/(2N).”
THE CORRECTION: “Step 2 (fixation probability): Each neutral mutation IN A REPRODUCING INDIVIDUAL (emphasis mine) has fixation probability 1/(2Ne). Each neutral mutation in a non-reproducing individual has fixation probability zero (not 1/N, as Vox’s “algebraic error” analysis wrongly assumes).”
Kimura’s celebrated result “k=μ” (albeit solely for neutral mutations) now follows immediately.
For historical context, two (relatively recent) survey articles by Masatoshi Nei and colleagues are highly recommended: “Selectionism and neutralism in molecular evolution” (2005), and “The Neutral Theory of molecular evolution in the Genomic Era” (2010). In a nutshell, Kimura’s Neutral Theory raises many new questions — questions that a present are far from answered — and as Nei’s lively articles remind us:
“The longstanding controversy over selectionism versus neutralism indicates that understanding of the mechanism of evolution is fundamental in biology and that the resolution of the problem is extremely complicated. However, some of the controversies were caused by misconceptions of the problems, misinterpretations of empirical observations, faulty statistical analysis, and others.”
Nowadays “AI-amplified delusional belief-systems” should perhaps be added to Nei’s list of controversy-causes, as a fresh modern-day challenge to the reconciliation of the (traditional) Humanistic Enlightenment with (evolving) scientific understanding.
Another strikeout. He removed the non-reproducers twice, because he doesn’t understand the equation well enough to recognize that Ne already incorporates their non-reproduction, so he can’t eliminate them a second time. This is the sort of error that someone who knows the equation well enough to use it, but doesn’t actually understand what the various symbols mean is usually going to make.
Kimura remains unsalvaged. Both natural selection and neutral theory remain dead.
DISCUSS ON SG